Metabolic
[ Muscle ]
Disorders
|
|
We use this title very loosely, as it really does not describe any single narrow conceptual entity. Too loosely, it could almost be the only category outside trauma. Essentially, having invested mental energy in learning the central metabolic pathways we want to focus on relevance to that schema - on defects entering, within and leaving that metabolic chain. Defects do alter movement but do not limit disability to muscle. Neurologic impairments may be severe. Bodily organs such as the liver may be prominent in their involvement.
Carbohydrate Metabolism
Diabetes Probably the first carbohydrate metabolism disease mentioned ought to be
diabetes. Although it is a bit peripheral to pediatric orthopedics, how can we ignore such an important subject? Glucose does not just float in and out of
cells. It could, but doesn't. Why? Because the body fences it in by phosphorylating it. Adding a phosphate to glucose effectively prevents it from
entering or exiting a cell unless it is invited. Blood levels of free glucose and intracellular levels are precisely regulated by a wide range of mechanisms.
Probably the first carbohydrate metabolism disease mentioned ought to be
diabetes. Although it is a bit peripheral to pediatric orthopedics, how can we ignore such an important subject? Glucose does not just float in and out of
cells. It could, but doesn't. Why? Because the body fences it in by phosphorylating it. Adding a phosphate to glucose effectively prevents it from
entering or exiting a cell unless it is invited. Blood levels of free glucose and intracellular levels are precisely regulated by a wide range of mechanisms.
Certain cells in the pancreas produce insulin, a messenger. Insulin is a small protein key that fits a cell membrane switch which turns on an intracellular cascade of actions that process glucose. Proteins within cells handle glucose by degrading it as fuel or by linking it in long strands for storage to be later unlinked when needed.
The two big sub categories of diabetes are 1) insufficient insulin (loss of the main messenger) by any of several mechanisms (such as autoimmune destruction of the key manufacturing sites) and 2) a goobered target mechanism that does not respond properly to insulin.
Insulin dependent diabetes (IDM) (wherein the insulin source is impaired) accounts for under 20% of the cases, but this varies very much by population mix. In certain pancreatic cells, rising glucose in the blood triggers a release of already formed and stored insulin into the blood and an increase in manufacture of insulin. Absent the insulin messenger to signal glucose supply while leaving the run off pathways intact is more prone to produce ketosis and coma. Somehow, the genetics of this form alters the insulin forming cell's surface chemistry such that it is prone to autoimmune cell destruction.
Fat requires some carbohydrate to get into and keep the citrate cycle going. Lacking the ability to generate oxaloacetate directly (to prime the cycle), the metabolism of fat piles up unfinished fat metabolism products - ketones. These ketones are not mere waste as once thought, but fuel. The heart uses the ketoacids as major fuel. The brain normally runs on just glucose, but can adapt to utilize ketoacids. Nevertheless, ketones, signify fatty acids burning without sufficient glucose to balance the citrate cycle.
Noninsulin dependent diabetes, (NIDM) the other form of diabetes , much more prevalent, involves relative blocks in the pathways of glucose metabolism. Insulin triggers the liver to release glucose from glycogen stores (and release amino acids as well). The cells, if stimulated enough, may form transport units to further enhance cell intake of glucose. Glucose, in excess of that needed by the cell, can be strung together to form a starch-like storage polymer (glycogen) or the excess glucose can be sent down the anerobic fuel pathway toward fat. Problems in regulating these steps are the basis for acquired diabetes. Once rare, there is a nearly epidemic rise in this type of diabetes in children in the United States . The proximate causes are known. Statistically, the smoking guns are television, computers, and obesity. The combination of lesser time spent at physical activity and increased weight (to say nothing of the kinds of foods pushed by advertising and fast food chains) clearly correlate.
The liver and the pancreatic cells that make insulin are not the only organs in direct control of glucose. Certain cells of the pancreas also make glucagon, another protein messenger that tells - especially the liver - to break down glycogen (the storage polymer form of glucose) and release glucose into the blood. The adrenal glands release epinephrine, a small signal messenger that tells cells, especially muscle cells, to increase intake of glucose and ready the break down pathways. The single most orthopedically relevant damages are obesity - and later - vascular compromise from poor diabetic control.
|
|
Glycogen Storage Diseases
|
|
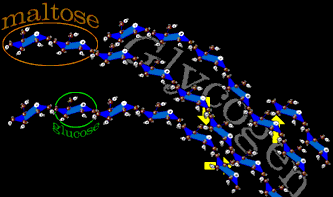
The animal form of starch is glycogen. If a single sugar molecule (glucose) were a bead, two beads stuck together would be maltose. Glycogen would be costume jewelry - not just long arcing chains but complex branching chains. Glucose, alone, is very water binding. A storage of soluble glucose would attract huge volumes of water. By tying up all those side attractions with other glucose molecules, billions of units can be stowed without becoming a water sponge. Also by branching the long strands, more glucose ends are available for rapid glucose release. Storage is at low cost as nearly 98% of the energy is conserved for usage. The yellow arrows show side to side linkages that are the branching points.
Remember hexokinase? (the protein shaped like a mouth that takes in a 6 carbon sugar and an ATP and transfers a phosphate from ATP to the sugar)? The liver has its own special version (a glucokinase called glucose-6-phosphatase or G6Ptase) which is specific to glucose (rather than all the similar 6 carbon sugars). This enables the liver to help regulate blood glucose levels. So, if glucose were to pile up behind a defective liver specific glucokinase, effects would most likely be evident in the liver itself. It is so as will be discussed below.
There are enough different defects of these complex pathways already confirmed to make the head spin. We will use a conceptual grouping as found in Robbin's Pathology: Those that primarily affect the liver, those that primarily affect the muscle, and the rest.
Primarily Liver :
Example: Von Gierke's Disease is the first type of sugar path blockage. It is from defective Glucose-6 -Phosphatase, a liver specific hexokinase. It is an essential protein needed to get the phosphate off of glucose-6-phosphate. So tagged, the glucose cannot leave the liver cell. Highly charged phosphate is like house arrest for glucose as it cannot pass freely through the lipid layer of the cell membrane . It backs up and is diverted into glycogen. The liver bloats with glycogen as the blood goes wanting for free glucose. The liver also tries to burn off excess glucose generating high levels of pyruvate and lactate. Peripheral mechanisms turn to protein and lipid for fuel generating high uric acid levels and lipids in the blood. The kidney, having some similar protein makeup, also develops glycogen deposition. Growth is stunted.
Primarily Muscle :
Muscle uses fat for energy but gets quick energy from glucose released from local muscle glycogen storage as well as from blood glucose. Glycogen is not just strands of glucose. It is highly branched and looks like a tree. Glucose is linked end to end, but every so often (ten sugars or so) a glucose is hooked side to side creating a branch. It takes a special enzyme to break the branch linkages.
The enzyme that nibbles glucose off the glycogen structure cannot nibble past a branch. Therefore access to glucose might be blocked if the glucose nibbler is defective or if the branches cannot be undone or if the storage form, glycogen, can't be made in the first place.
Example: In McArdle's Disease, the enzyme (named "muscle phosphorylase") is missing. It nibbles glucose molecules off of glycogen by adding a phosphate on the glucose at the break point (on the first rather than 6th carbon) creating glucose-1-phosphate (G1P). The G1P is readily converted to the key G6P which enters the anaerobic fuel pathway. Whereas exercise normally generates lactic acid (at the end of the anaerobic breakdown of glucose0, lactic acid is missing in McArdle's patients, absent the availability of glucose. The muscle burns creatinine phosphate instead with accumulation of ADP. High levels of ADP causes cramps.
Normal muscle can use energy from fat or from glucose. It takes muscle a while to switch over to pure fat metabolism (burning fatty acids from the blood). Typical is exercise - cramps - relief - then stiffness. Jaw and finger stiffness may even occur. Commonly the fingers cannot ungrip (extend) after strong clenching activity. Weakness and exhaustion are prominent after exercise. Muscle wasting is more prominent in the pectorals than in the muscles around the pelvis.
Less Specific - the rest :
Example : Pompe's Disease. Lysosomes are intracellular membrane sacs which store certain enzymes, usually enzymes that digest stuff. If the glucosidase of lysosomes is missing then lysosomes bloat with glycogen. Bloated lysosomes, being broadly distributed, will affect more tissue types including heart and skeletal muscle, as well as liver. Very low muscle tone with a very enlarged heart is followed by respiratory failure. This is the typical version of the more distributed defect type.
Under each of these three categories are an array of other disorders, some with syndrome names, and some just named after the enzyme or gene location identified.
Fat Metabolism
Fats are built up in units of two carbons and utilized by splitting off pieces of two carbon size from the fatty acids. First, the fatty acids get chaperoned to this process by Coenzyme-A. This coenzyme is located on the mitochondrial outer membrane.
Small fatty acids can be passed on and into the mitochondria to be metabolized. However larger fatty acids - "long chain" fatty acids cannot pass into the mitochondria without assist. Carnitine takes the long fatty acid from the coenzyme-A and passes it through a very large protein (translocase) structured as a passage door in the mitochondrial membrane. Once inside, the carnitine releases the long chain fatty acid to another Coenzyme-A located inside and returns to the outside. One metabolite of the citrate cycle inhibits fat entry into the mitochondria (by the carnitine mechanism being inhibited). This is a regulatory step.
If fat becomes the predominant fuel, oxaloacetate becomes scarce as little of the fat degradation makes it all the way around the citrate cycle. The acetoacetyl build up shifts to acetoacetate and then acetone and Hydroxybutyrate (these are called ketones). The heart and kidneys burn acetoacetate as regular fuel, the brain can use it when glucose is severely low. Interestingly these ketones slow the release of fat from fat cells. Therefore, glucose starvation can encourage fat to stay in fat cells.
Provision in the diet of synthetic fats which have an odd number of carbons ( odd chain fatty acids) will create a ketosis due to the left over odd parts. Ketosis has been found to be a powerful means to depress seizure activity.
|
|
|
|
There is no storage mechanism for amino acids. If there are unused amino acids, then they are broken down. The amino acids all share a common
,OH
NH2-C-C=O
|
R <= where R is a side piece that varies for each amino acid.
The nitrogen part is the 'amino' which gets chopped off and winds up as ammonia. The remainder of the structures wind up as pieces which can enter the citrate cycle (different amino acids enter at different places depending on the nature of the -R piece. The final fate of ammonia ( NH4+ ) is to wind up as urea by way of a cycle that takes in nitrogen and sends out urea ( urea cycle) :
NH2
|
C=O
|
NH2
Disorders in the enzymes that dispose the amino group as urea will cause ammonia to build systemically and cause coma and death. Reactions to pull off nitrogen and create a disposable urea take place in part in mitochondria and inpart in the cell cytoplasm. Thus the urea cycle is linked to the citrate cycle.
Vitamin B12, a complex multi ring molecule with a cobalt atom dead center, is used as a tool by a protein whose job is to break down certain amino acids. It also is the means of making deoxyribose from ribose (in DNA, the D=deoxyribose. in RNA, the R=ribose). It is also used in methyl donor reactions.
In methyl malonic acidemia, large amounts of this substance appear as the baby slips into coma. Half of the cases will respond to B12 , in large doses, as the defect is that of getting the B12 to the needy protein that uses it. Half of the cases will not respond as the defect is that the protein itself is missing or defective. Very odd diets which avoid the amino acids which require that enzyme, treat the disease. An example of this is shown in the muscle transfers section.
Hundreds of specific diseases, relevant as they are, were not mentioned here. The scope is too wide and this discussion too general. Use this information as a skeleton of the basics on which to hang the specifics supplied by your doctors.