Muscular Dystrophy
Surely, if ever there were a disease that pitted mankind
squarely against nature - this is it.
 |
There are kinds of muscular dystrophies. This is the nastiest and the one we know best.
Depending on who is counting and how, about 1 in 5000 male children will develop this disease. As DNA identification methods are already showing that there are a variety of ways for the gene to be defective, we may well find variants of DMD that were not so called by the methods that have gone before.
The clinical essence is simple enough. Somewhere after 1 year old and before 6 years old a healthy male child begins to fail. It is subtle, at first. But soon enough weakness becomes apparent.
A tendency to toe walk - which was not there before - evolves.
male child begins to fail. It is subtle, at first. But soon enough weakness becomes apparent.
A tendency to toe walk - which was not there before - evolves.
Getting up from the floor becomes an effort. Rather than an enthusiastic spring to the feet a series of bodily repositionings occurs with hands pressing against legs to assist getting to an upright posture.
The muscles that seem most weak do not seem to be smaller or softer. In fact the calves seem husky. They downward pointed feet do not produce a strong push. They do not push beyond their own posture at rest with any strength. They are weak, but stuck.
 What is happening?
What is happening?
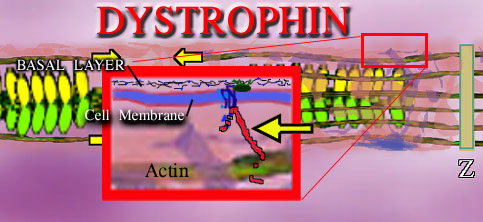
Each muscle cell is bounded by its cell membrane - a three layer affair of phospholipids with many variations. Outside the cell is a mat of fiber, of very strong sort, called the basal layer. This tough fiber interlocks with itself to form the strong connective fiber between cells. It is made of a protein called collagen (type 4 in this case). This basal layer holds sheets of cells together. If forces are to be transmitted to the outside world from within
the cell, somehow you have to pull on this connective tissue between the cells.
Four loops of a protein are embedded in the cell membrane and like four loops of stitches holding a button on a shirt, these loops hold a triple fish hook like protein. These connecting proteins (glycoproteins) are reinforced within their structure with sugar polymers and are very strong.
The purpose of this cell membrane molly which fixes into the fiber outside the cell, is to support the actin inside the cell. The protein support rod which latches onto actin is called DYSTROPHIN. It is dystrophin which is missing in whole or in measure in the Duchenne and related dystrophies. In Duchenne the absence of dystrophin is profound. In Becker dystrophy the absence is partial. The degree of relative loss is the difference between severe Becker's dystrophy and mild. Other dystrophies have defects in the glycoprotein components.
Interestingly, dystrophin is a HUGE molecule that has a long reach - think of it as filler. Within that long reach are sequences or sub segments that can be and are used for other purposes in other tissues. So dystrophin is a montage of other molecules. Thus defects of those sub segments may reflect in abnormalities within the eye, brain and other tissues. Because dystrophin is such a very large long molecule of many amino acids in a row, there can be a huge variety of alterations in that length by code mishaps. Some don't matter. Some maybe throw it off a wee bit. Other locations ruin the whole purpose of the molecule. So right there we see that there can be a large array of severities and exact DNA abnormalities associated with this disorder.
Exactly how dystrophin absence kills cells is unclear, but cellular fiber holding cells together in stress zones is known from other diseases which have different fibers amiss. In epidermolysis bullosa, missing keratin fibers result in cells pulling apart. Skin peels as if burned.
This is deeper than skin, but the forces are those of the outside world and of actin being pulled by myosin with no cell envelope support. It may very well just pull holes in the cell. Early chemical tests for this disease simply detect muscle cell proteins which ought not be floating around outside the cell. An ATP regenerating protein which helps creatinine get phosphate to ATP (creatinine phosphokinase) has no place outside the cell. Its presence in the blood is evidence of muscle cell rupture.
Is rupture the actual damage? Don't know, but probably. The involved areas become abuzz with the migratory cells used by the body to cart away damage. Inflammatory cells are inflammatory because they have to first degrade spilled cell substances. That degradation requires digestive enzymes to chop the bigger stuff into smaller forms that can be disposed. Scar - dense collagen - forms in the damage areas.
 |
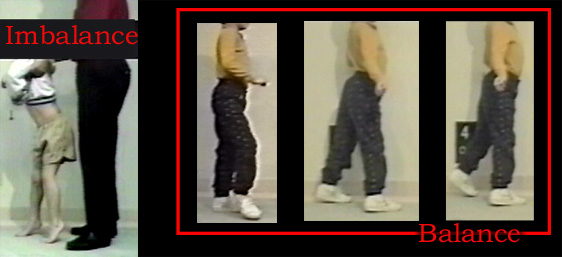
Thus we have less muscle (weakness) as damaged muscle is replaced by scar (contracture as scar is unyielding ). The inflammatory residua more than offset loss of muscle, so that volume may actually increase. Normal standing balance would have the center of mass of the body pass through the plane of the hips and knees and be centered in the mid-foot (forward the ankles). This is a very precarious balance. With muscle control - precarious is good in that we can easily move and quickly if need be.
In DMD the hip extensor gluteus maximus is weakened and the hip would tend to flex. To maintain balance upright, absent hip extensor strength, the body shifts backward to place the center of weight behind the hip. That would tend to extend the hip that would otherwise fail in flexion. That the weak hip extensors allow the pelvis to forward roll, arching the back is even more so exaggerated to get the weight sufficiently behind the hips. This backward bending of the spine is called LORDOSIS.
The quadriceps are weak. A back knee places body weight in front of knee center allowing the knee to mechanically lock. The downward angle of the stuck foot (calf is stiff and contracted) helps drive the knee back, making up for absent quadriceps (front thigh) power. That balance is very narrow and when the foot goes too far down, the backward lean of the leg goes beyond that which can be controlled (outside the control arc - very narrow one at that). The toe weight bearing is simply too forward to allow hip and knee locking to both occur. At this point parents discover shoe heel lifts or wedges, or serial casts are done etc. to get the support just forward the ankle.
 |
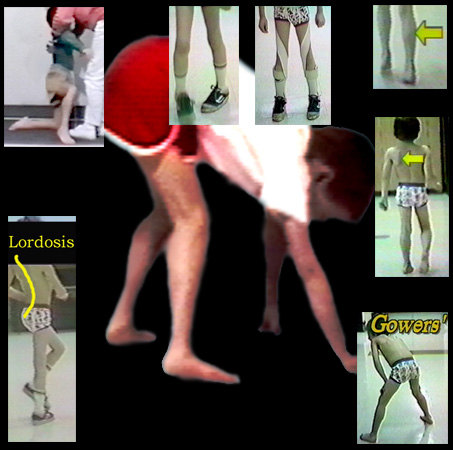 At 1:00 we see calf swelling. This is not muscle but scar. At 2:00 we see shoulder winging which shows that the scapula muscles are also affected. At 5:00 the
classic Gowers' posture of getting to standing by means of the hands. At 7:00 we see the backward arch of the spine in walking (lordosis) which places the body mass behind the hip joints and prevents
then from flopping into flexion. At 11:00 we see weakness in the arms and shoulders so sever that even hands can't assist getting up. It is difficult to pull the youngster up by the hands or arms as the body falls through
unsupporting limp shoulders. Straddling 12:00 we see the inward rotation that is a consequence of the fact that the gluteus maximus hip extensors are also the main outward rotator muscles. Straps which
return the straight ahead rotation of the legs make walking look better, but reality is that the function is nearly the same unless overt tangling of feet is occurring. That is unlikely as the legs get
wider and wider apart as the muscles called "tensor fascia lata" become contracted and a wide base ensues.
At 1:00 we see calf swelling. This is not muscle but scar. At 2:00 we see shoulder winging which shows that the scapula muscles are also affected. At 5:00 the
classic Gowers' posture of getting to standing by means of the hands. At 7:00 we see the backward arch of the spine in walking (lordosis) which places the body mass behind the hip joints and prevents
then from flopping into flexion. At 11:00 we see weakness in the arms and shoulders so sever that even hands can't assist getting up. It is difficult to pull the youngster up by the hands or arms as the body falls through
unsupporting limp shoulders. Straddling 12:00 we see the inward rotation that is a consequence of the fact that the gluteus maximus hip extensors are also the main outward rotator muscles. Straps which
return the straight ahead rotation of the legs make walking look better, but reality is that the function is nearly the same unless overt tangling of feet is occurring. That is unlikely as the legs get
wider and wider apart as the muscles called "tensor fascia lata" become contracted and a wide base ensues.
The disease goes on. Trunk muscles may fail and allow spinal forward and side to side curvature which progresses with maddening speed. Because of the joints on the back of the spine are arranged in oblique overlapping manner, back extending lordosis not only keeps the diaphragm from being pushed up by intestines etc, but lordosis also locks the side to side flexibility against scoliosis. One of the main reasons for scoliosis soon after walking is lost is that the seated posture is not in lordosis. Therefore seating ought to try to mimic the lordosis seen in walking. However, if scoliosis does occur, braces ought NOT be used. The reason is that they always fail and not only fail but delay surgical treatment to a time when the child cannot survive it. Even a small scoliosis - if it is to be treated at all - requires prompt spinal surgical stabilization.
Some centers use magic numbers such as 30o to mark the need for surgery. However, that is almost irrelevant as any progressive curve will quickly attain and pass that mark or any other mark. If the curve is seen to be progressing, it's time. Earlier surgery is simpler, more effective and far less risky.
How is survival figured in this? Well, for one thing, the ability to breathe - diaphragm and rib muscles - is getting poorer. The ability to self support a surgical aftercare may be passed. But too, the heart muscle may be compromised. Indeed, the only thing keeping some of these children alive is their total motionlessness. The heart may be 99% replaced by fibrofatty tissue and look like very cheap bacon - just small thin strips of muscle scattered here and there.
Clearly, the answer to this disease is not surgical. Surgery helps prolong some activity but does not alter the systematic problem.
That some children are overly sensitive and cry out in pain at any manipulation suggests that nerve, too, may be involved. Some - only some - lose mental proficiency to a degree. Recent findings of a form of the dystrophin protein complex in various neural tissues supports the notion that the basic defect is predominant in muscle but not confined to muscle.
Let's look at the peculiarities of walking and introduce a few concepts that seem to have missed everybody's attention. This discussion is about efficiency and energy, also discussed elsewhere.
At the origin of walking is intracellular myosin doing a tug-of-war on actin. That costs energy. ATP and other energy molecules get spent in the molecular activity of muscle action. In muscular dystrophy there are two graphs to follow. One is that of energy delivery as O2 and fuel delivery require the lungs to work and the heart to pump. The other is efficiency of walking. The graph is energy demand (fuel usage) as walking becomes compromised. What stops walking is NOT just mechanical inability but energy insufficiency. Thus the fact that certain steroids have benefitted the pumonary and probably also the cardiac picture explains the claimed prolongation of walking. You do not need to assert any effect of those medications on the muscles of the legs, just maintenance of the energy delivery mechanism.
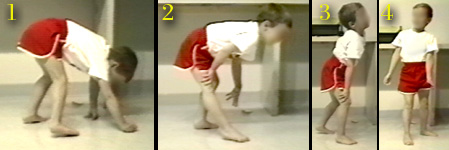 |
The Gowers sequence of getting erect points out the profound weakness of certain muscles. But once the child is up, standing may be at fairly low energy cost bys substituting joint locking tricks for stability.
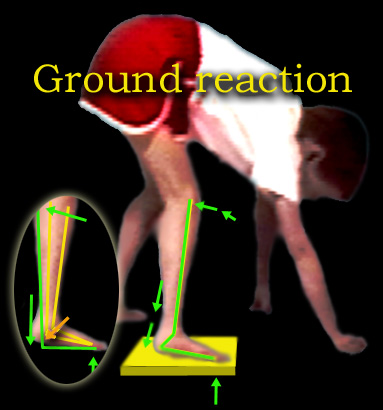 The first item is that the ankle becomes STIFF. It also points down a bit. When the toe is on the floor, the heel is - at first - not on the floor. As weight is
applied, the stiffness of the ankle drives the tibia (shin) backward and thereby locks the knee.
The first item is that the ankle becomes STIFF. It also points down a bit. When the toe is on the floor, the heel is - at first - not on the floor. As weight is
applied, the stiffness of the ankle drives the tibia (shin) backward and thereby locks the knee.
The knee extends just a bit beyond straight and like a baby carriage hood mechanism, needs no further action to stay straight. Thus foot ankle stiffness substitutes for absence of knee extension power (quadriceps
weakness). If the knee has a flexion contracture of its own that prevents this overshoot, then the stability mechanism fails.
a baby carriage hood mechanism, needs no further action to stay straight. Thus foot ankle stiffness substitutes for absence of knee extension power (quadriceps
weakness). If the knee has a flexion contracture of its own that prevents this overshoot, then the stability mechanism fails.
The inability of the knee to fully extend is usually a consequence of prolonged sitting. The hamstrings and the joint itself become resistive. Hamstring shortening furthers inward rotation of the legs.
Also on the stability side is the requirement that the tibia not be pushed too far back. If that happens, then the body must lean forward to have center of mass over the feet. That forward lean at the hips is called "jack knife" posture. There is a very important misconception floating arount among PTs and others who should know, that the jack knife hip flexion is a hip flexion contracture. Further the misreasoning is that the child goes up on his toes to offset that hip flexion contracture. Not true. Staying up on the toes is a way to not require jack knife posture when the ankle equinus has gone too far. What's the big deal? Semantics? No. There are those who advise hip flexor (psoas) releases or lengthenings for this!! Wrong. The hip flexors are the most important energy generators that we have in walking. Further, they are major sitting stabilizers of the spine. Weakened psoas disturbs sitting control. Further still, psoas muscles produce lordosis. Lordosis is protective as described above.
 The other key to continued walking is efficiency. The single most important facet to
efficiency is a supple swinging leg. The swing leg ought to swing like a pendulum. In weakness, the psoas and a bit of torso flipping action tosses out the thigh. The shin
segment follows as would any linked pendulum with no direct muscular driving action other than the momentum of the thigh above.
The other key to continued walking is efficiency. The single most important facet to
efficiency is a supple swinging leg. The swing leg ought to swing like a pendulum. In weakness, the psoas and a bit of torso flipping action tosses out the thigh. The shin
segment follows as would any linked pendulum with no direct muscular driving action other than the momentum of the thigh above.
In order to swing the leg forward, the foot-ankle unit cannot be too rooted to the floor. An
overly wide base or plantar flexed foot will glue the foot too firmly with the knee held back, impeding forward swing. Efficiency suffers at the expense of stability. Indeed, if a toe
walker has an operation that "gets the heels down" or a brace that does the same thing - but in too stable a configuration - then - efficiency suffers. High energy cost + low energy production = no walking.
An
overly wide base or plantar flexed foot will glue the foot too firmly with the knee held back, impeding forward swing. Efficiency suffers at the expense of stability. Indeed, if a toe
walker has an operation that "gets the heels down" or a brace that does the same thing - but in too stable a configuration - then - efficiency suffers. High energy cost + low energy production = no walking.
Wedges, or heel lifts, can be used to both test for the best degree of stability without over trapping. Often cowboy boots are worn because they supply this need.
 A downward hanging foot also requires a higher stride lest the toes drag the ground in swing phase. Toes that drag are more than annoying. They trip
and they cost energy as they dampen the swinging pendulum.
A downward hanging foot also requires a higher stride lest the toes drag the ground in swing phase. Toes that drag are more than annoying. They trip
and they cost energy as they dampen the swinging pendulum.
Walking on toes offsets this disorder as the height is offset by the other side. Getting only one foot "down" and not the other will disturb gait significantly as the opposite side no longer compensates.
But weakness is weakness. As it progresses, these subtle compensatory tricks cannot be managed across the three levels of the leg. Extremely fine tuning of the control arcs is required to allow the walking mechanism to resume as is seen below (before and after percutaneous surgery under local).
 |
This is as good a place as any to instruct the basic differences between the two schools of surgical thought. The child above had an attempted larger surgery aborted when cardiac arrest followed anesthesia inhalation. Fearful of surgery, the alternative method (percutaneous lengthenings under local) was opted only after walking failed altogether. Percutaneous methods are best suited to the point of failure - whether bracing is used or not. Comparitive data for this method is built in, as each patient is his own control. Having lost walking, he either resumes or not. The key is absence of fixed knee contractures.
mpg-1 =>
|

At this point there is a Gowers' sign but gait looks nearly normal. The "wheel" configuration of leg to leg transfer is seen (right toe to left heel). The walking base is narrow. Postoperative the patient will look good because preoperatively he is good. But does this really extend walking time. They claim it does. No data, though, as we can only guess - without controls - how long these kids would have walked. Complications at this stage are harder to bear than later. Tough call.
The third school is not surgical. The English tradition uses extensive and clever bracing early and freely. It is not to be criticized as it does serve its population when the people who do it are proficient at it and timely. Bracing in the USA is willy nilly. You write a prescription and half of the time you have no idea what you are going to get - or when!!! So surgical methods have rooted in firmly.
The rest is just details. Some cases present earlier, some later. The degree of severity does vary. Some are so mild as to even question the diagnosis. As the disease is rare in females, we have long known that the gene defect must be on the X chromosome (XX=female, XY=male, it would take both X chrosomes to be defective for a female to have DMD. It has happened. ) A consistently less severe form is known, called Becker Muscular Dystrophy.
Becker dystrophy disrupts walking less and later, with many walking well past 20 years of age and without severe general deteoriation. Intellect loss and heart degeneration is not prominent.
Limb Girdle Muscular Dystrophy is probably not, in a clinical sense, a single entity as many cases of spinal muscular atrophy wind up with this label - though less so nowadays. Nearly twenty different molecular variants were loggen in last time I looked. So this group is a group by way of how it differs from DMD so to speak. The group is characterized by predominant proximal (nearer the body) involvement so that shoulders and pelvis are disproportionately weaker than the rest. It is not limited to males. A female with MD probably belongs in this group (or the next). Severity can be similar to Duchenne's or be quite mild. The scope of involvement is less stereotypic than Duchenne's, also suggesting that this may be a mixed cause group. Looking at the many pieces involved in the linkage of actin to the basal layer, this should be no surprise.
Fascioscapulohumeral Muscular Dystrophy is inhereted as a dominant unlike the Duchenne (even though Duchenne described it first, two other names are usually tied to it as one name for two syndromes would be confusing. Thus : Landouzy - Dejerine Dystrophy). The distinctive feature is facial involvement - though shoulder girdle weakness and winging may be seen first. Lifting the arms out from the body to the side will ride the shoulder blades way up so that they can be seen from the front above the normal neckto shoulder contour. Some cases show early signs in childhood, but most come to light in adolescence and adulthood. Females are seen in this group. Many are misdiagnosed, however, in both directions as myasthenias can look similar. Current diagnostics are making the groupings truer to the root causes. At least a chromosone 4 locus has been found as autosomal dominant (90%ish). The other 10%- stay tuned.
Congenital Muscular Dystrophy is the one variant that tends to improve. The stigmata may be very dissimilar as the early onset can cause other syndromes. Arthrogryposis multiplex congenita, a birth condition that results when forming embryonic joints are not moved may result from anything that stops motion - neurologic, toxic, or muscular - whatever. At least some AMC are due to congenital muscular dystrophy. At least one form of CMD is a mitochondrial disorder.
That this group gets better - often - musclewise - brings up a common theme in pediatrics. The process of getting built -embryo to fetus to out- takes place in the womb. The process is very demanding metabolically and enzymes are highly specialized for that environment. That includes hemoglobin - there is a different hemoglobin in the fetus because O2 has to be gotten from mother's hemoglobin which has to want to give it up. The fetal hemoglobin needs greater affinity for the oxygen, yet not so much it won't give it over to the baby. Once born, this middle of the road version is less good as now it must get oxygen from the air in the lungs. So a fetal protein gets shut down as the adult form kicks in.
WHAT IF - the adult form is defective? Well there is an anemia category for missing adult form hemoglobin. The generality, what if any enzyme has two forms and the adult form is just not there or defective. Then something that was working at birth will fail. Conversely, if the fetal form is substandard but the adult form is OK, then the disease seems to get better with age.
Is this the case with certain muscular dystrophy types? The congenital form improves and the childhood forms worsen. I don't know. Just a guess.
Interestingly, a blood pressure drug (Losartan) has the ability to slow breakdown in MD as well as another inhereted disorder (Marfan's). It has to do with regeneration of cells blocked by a certain factor that was being followed in high blood pressure. Don't sweat the details - this is all mice at this point - but just know that directed research isn't the key. Broad research is. Surprises come up in odd contexts. It's all connected.
There is an abbreviated way to view this disorder. Consider that the contractile fibers of muscle need to link with the force carrying connective fibers outside the cell. Then a fiber needs to go from inside the muscle cell to outside and join the connective tissue that eventually becomes tendon. This linking protein is the longest protein in the body (so it has a long code, 3 DNA thingies to each amino acid, and thus has a lot of places to go wrong or at lest to vary. Some miscodes place odd amino acids in place of correct ones but span the area well enough to still work - maybe a bit less well - but OK. Where the long fiber (we are talking about dystrophin) penetrates the cell membrane there is a cluster of smaller molecules that you can think of as a grommet that transition the penetration. Defects in these smaller proteins and helpers also can lead to cell damage and look a whole bunch like Duchenne's - in fact they are included in that term.
So it turns out that any of the many possible alterations along that long dystrophin molecule can cause the clinical picture we call Duchenne Muscular Dystrophy with muscle cell break-up. So can defects in those transition proteins - thus the occasional oddball test negative cases that didn't test these. Well in U. of Illinois the folks there have noted that one of those helper molecules (integrin) can be supplied when that one is abnormal and help (mice). But they also found that integrin in excess can also stabilize some of the dystrophin anomaly cases. Wanna bet those are the ones where the dystrophin is abnormal at the place where it interacts with integrin - make a better grommet for an iffy dystrophin transition area???? Anyway, this is where research is at. Looking for cures and maybe work-around solutions.
OK. Here's another. We know that the exit point of that long dystrophin fiber from the cell needs to be secure. If not? It leaks. Specifically Calcium ion leakage does the big cellular damage. Folks in NYC at Columbia noted that heart failure has a similar problem. A drug used to treat the heart for such leakage is being proposed for Duchenne's. Makes sense. It is a delaying tactic, not a cure, but we need more such ideas.
Did you know that cells have a suicide mechanism? Yup. There is a mechanism that tells a cell to self destruct. A gun like that needs the safety keep on. A protein that signals this path to cool it, may be deficient in certain 'limb girdle' types of muscular dystrophy. So muscle cell death, by whatever means gives the syndrome its look. The causes are many and they determine the scope and speed of the problem. Stay tuned.
There are still others: Oculopharyngeal, Distal, and Emery-Dreifuss to name a few. The thing is to follow the genetic tracking sites for the details as they are chasing many leads, much is still too esoteric to say aha!
 |